作者:Xian Su、Haixue Wang、Tao Wang(国家药品监督管理局药品审评中心);Nan Zhao、Yimin Cui(北京大学)
由于过去十年的监管改革,中国新药的临床开发和监管审查过程发生了重大变化。监管改革的目的是鼓励开发创新产品并确保病人能及时获得这些治疗,尤其是针对罕见、严重或危及生命的疾病的药物。为了评估这些流程更改的影响,我们分析了2010年至2020年中国批准的创新药物的试验性新药(IND)申请和新药申请(NDA)的数据。
2010至2020年新药发展趋势
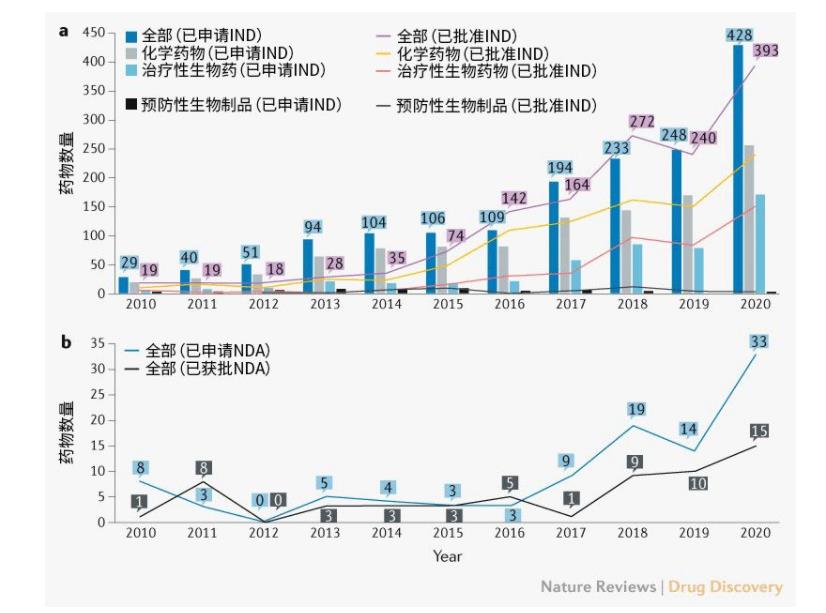
首次IND申请数量大幅度增加。2010至2020年期间,共有1636个创新药物提交了首次IND申请,平均年增长率达32%(图1a)。其中,1410个药物(86%)来自689家中国公司。值得注意的是,与2016年相比,2017年创新药物的首次IND申请数量增加了78%,其中化学药物和治疗类生物产品分别增加了60%和159%。
图1 | 中国创新药物年度INDs和NDAs数量。a | 创新药物的每年度试验性新药(IND)申请数量。根据药物类型,这些药物被分为三大类。b | 创新药物的年度新药申请(NDA)数量。IND申请和NDA的数据按首次提交申请的年份进行分类,已获批的IND申请和NDA的数据按首次获批的年份进行分类。
在1466个完成的首次IND审查中,有1404个(96%)获得批准。被拒原因主要包括申请后研究信息缺乏导致沟通不畅、药物临床效益风险比不合理以及违反临床诊断基本原则等。首次IND批准的创新药物数量每年增长35%,其中2016年首次IND批准的创新药物数量显著增加。
2015年7月,中国出台了监管改革措施,缓解了申请积压,鼓励了药物创新。与此同时,创新药物的首次IND申请和批准数量大幅增加。
获批的创新药物NDA数量虽少,但在不断增长。2010至2020年期间,共提交101种创新药物NDA,58种创新药物获批。近年来,提交和批准的NDA数量普遍增加(图1b),58个获批创新药中有42个(72%)自2015年7月之后获批。
在已完成的66个药物首次NDA审查中,有58个(88%)获得批准。未获批主要原因是药物或临床研究设计存在重大缺陷、不同开发阶段使用的研究样本不一致以及审查中发现的临床试验数据不真实。
在58个获得批准的创新药物中,37种药物为通过中国国家药品监督管理局(NMPA)批准的新分子实体(NME),而另21种药物为在国外批准或销售的药物、传统疫苗、血液制品、细胞因子等(补充表1)。而且,有14种药物(24%)为孤儿药,其中13种药品符合美国食品药品管理局的孤儿药标准。此外,40种创新药物(69%)获得了优先审评,其中35种(88%)在2015年7月之后获批(补充表1)。数据表明,2015年8月推出的优先审评等监管改革措施鼓励了机构创新。
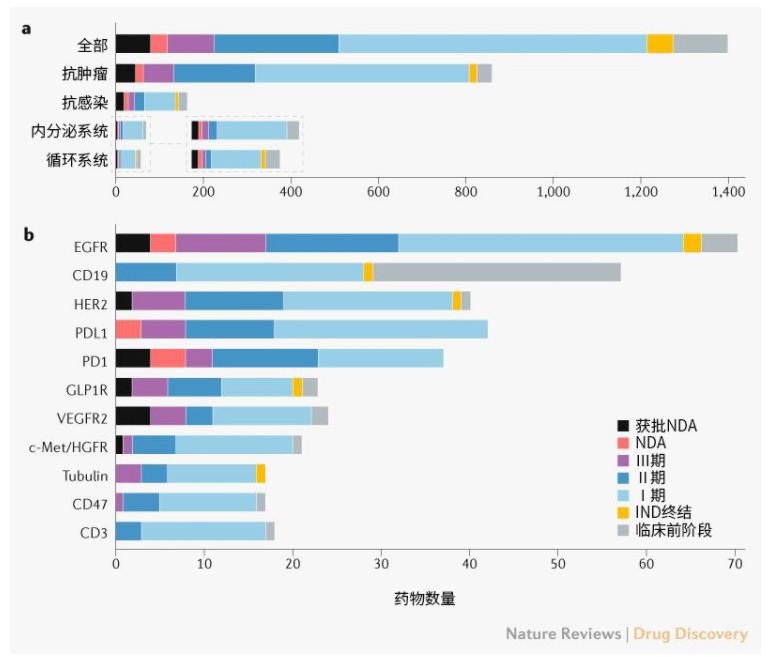
肿瘤学是创新药物的主要领域,大多数药物处于早期开发阶段。首次申请IND获批的创新药物的主要治疗类别是肿瘤学(864,62%)、感染(166,12%)、内分泌疾病(72,5%)和心血管疾病(60,4%)。2016年获批首次IND申请的肿瘤药物数量显著增加,增长率为133%(补充框1)。
在首次申请IND获批的创新药物中,705种药物(50%)至少进行到Ⅰ期实验,286种(20%)和108种(8%)药物分别达到了Ⅱ期和Ⅲ期阶段。大多数抗癌药物(57%)进展尚未超过Ⅰ期(图2a)。
图2 | 创新药物开发阶段。a | 主要治疗类别。b | 药物靶点。药物可以针对单一靶点,也可以针对多个靶点。
中国主要处于快速跟进和渐进创新阶段,只有3种已获批NDAs的创新药物是首创新药(first in class):本维莫德(benvitimod)、甘露特钠(sodium oligomannurarate)和罗沙司他(roxadustat)(补充表1)。针对EGFR、PDL1和HER2的药物超过40种(图2b),其中针对EGFR有最多的批准药物(4种)和达到临床试验阶段的药物数量(64种)。具有相同靶标和适应症的多种药物会降低创新的效率。
IND和NDA的批准速度提高了,然而临床试验速度没有提高。如图3所示,改革后阶段(2015年7月至2020年12月)的IND获批时间比改革前阶段(2010年1月至2015年6月)缩短了414天(之后87天,之前501天)。同样,改革后药品的NDA审批阶段比改革前缩短了441天(之后483天,之前924天)。
图片
图3 | 2010至2020年创新药物获批阶段长度。IND审批阶段被定义为从首个IND申请提交日期到批准之日。临床试验起始阶段定义为从首个获批的IND申请提交之日起至该临床试验的第一名参与者入组之日。临床阶段定义为从IND申报之日起至新药申报(NDA)提交之日。NDA审批阶段定义为从首个NDA提交之日起至批准之日。***,P< 0.001;****,P < 0.0001。详见补充信息。
2018年7月,国家药品监督管理局将开始药物临床试验的审批流程调整为默示许可制度(补充框1)。就整体临床试验阶段的长度而言(之后2572天,之前2688天),两个阶段之间的差异不显著。而在IND获得批准后的改革阶段,将第一个参与者纳入临床试验的时间比改革前多了59天(之后328天,之前269天)(图3)。提高临床试验的效率是中国鼓励创新的一个重要改革方向。然而,这受到伦理审查效率、研究人员经验和临床试验资源等多种因素的影响,需要对国家临床试验能力进行战略升级才能解决。而且,目前相同靶标和适应症的药物集中在相同的临床试验阶段,加剧了对临床试验效率的影响。
展望
中国的监管改革促进了本土医疗创新。获批的IND和NDA数量趋势以及得到这些批准所需的时间表明,这些变化的影响总体有益。然而,具有相同目标的多个产品扎堆和效率不足的临床试验过程,将影响真正有意义的创新药物获得从IND到NDA批准的进展。
感谢Yue Yang和Xiaocong Pang在计划、文章初稿和数据分析方面的协助。
原文以Trends in innovative drug development in China为标题发表在2022年5月5日《自然综述:药物发现》上。本文来源:Nature Portfolio
本网站转载的所有的文章、图片、音频视频文件等资料的版权归版权所有人所有。如因无法联系到作者侵犯到您的权益,请与本网站联系,我们将采取适当措施。